What is Circle Sequencing?
Circle sequencing represents a pivotal progression in library preparation protocols for high-throughput sequencing, effectively expediting advancements within the expansive domain of genomic research. This innovative protocol diverges from the conventional linear sequencing methodologies, placing emphasis instead on the formulation of vigorous circular DNA templates derived from genomic DNA. This innovative mechanism provides heightened efficiency and notably improved error correction capabilities, positioning it as an essential asset within diverse scientific arenas such as cancer genomics, immunogenetics studies, microbial diversity exploration, and environmental sampling techniques. The following comprehensive discourse aims to delve intricately into the integrative principles, broadened applications, and profound benefits proffered by Circle-seq. Moreover, this disquisition will delineate the bioinformatics analysis particularly associated with this technique, underscoring its indubitable implication within contemporary genomic research methodologies.
Principle of Circle-Seq
Circularization of DNA Templates
Circle sequencing constitutes a paradigm shift in high-throughput sequencing techniques, providing a unique platform for genomic research. Central to this method is the fabricating of circular DNA molecules from genomic DNA. This intricate process commences with the fragmentation of genomic DNA into more manageable units, usually averaging 150 base pairs in length. Following this, the DNA motifs are phosphorylated and then denatured, priming them for the circularization process.
The act of circularization is catalyzed by enzymes, particularly CircLigase II ssDNA ligase, creating a lasso-like DNA configuration. Any excess, unstructured DNA is eliminated through an exonuclease cleaning action, ensuring the integrity of the remaining circular DNA templates. The end product is an efficiently prepared database for genomic exploration, epitomizing the transformative potential of Circle sequencing in genomics research.
Rolling Circle Amplification
Subsequent to DNA circularization, an approach known as rolling circle amplification (RCA) is enacted to multiply the circular templates. This involves attaching exonuclease-resistant random primers to the circular DNA and activating the augmentation procedure with enzymes like the Phi29 DNA polymerase. The RCA technique yields elongated concatemeric DNA chains containing numerous tandem repetition of the original template sequence, thereby considerably magnifying the DNA and boosting the sequencing signal.
High-Throughput Sequencing
Upon the generation of multiple copies of circular templates via RCA, these amplified forms are further subjected to sequencing. Generally, this sequencing is conducted through high-efficiency sequencing platforms that include, but are not limited to, Illumina MiSeq or HiSeq systems. Bi-directional or paired-end reads are subsequently created which enables the sequencing of both termini of the DNA fragments. This crucial step ensures the enhancement of the gross informational yield from each DNA molecule, thereby bolstering the precision and trustworthiness of the resultant sequencing data.
Take the Next Step: Explore Related Services
eccDNA Sequencing (Circle-seq)
eccDNA Methylation Sequencing Service
Discover More: Recommended Reads
Detailed Workflow of Circle-seq
The Circle-seq methodology comprises several intricate steps, exemplified herein, to ensure precise delineation of off-target loci:
DNA Fragmentation: Genomic DNA undergoes fragmentation via physical means such as sonication or enzymatic cleavage to yield fragments of predetermined lengths.
Purification: The fragmented DNA is purified to eliminate potential contaminants that might impede the circularization process, typically employing Agencourt AMPure XP beads.
Circularization:
a. Blunt-End Ligation: Purified DNA fragments undergo treatment to generate blunt ends, thereby facilitating circularization. This process involves the utilization of DNA repair enzymes in conjunction with blunt-end ligation.
b. Circular DNA Enrichment: Circular DNA entities are enriched from the amalgam of linear and circular DNA molecules. This phase frequently entails exonuclease treatment to degrade any residual linear DNA fragments, leaving behind circularized DNA moieties.
In Vitro Cleavage and Sequencing Library Preparation:
a. In Vitro Cleavage: The enriched circular DNA is subjected to the genome-editing nuclease (e.g., CRISPR-Cas9) in vitro. The nuclease induces double-strand breaks (DSBs) at both target and off-target sites within the circular DNA.
b. DNA End Repair and A-tailing: The cleaved DNA fragments undergo end repair to generate blunt ends, followed by adenylation of the 3′ termini, rendering them amenable for adapter ligation.
c. Adapter Ligation and Amplification: Sequencing adapters are ligated to the adenylated DNA fragments, subsequently amplified via polymerase chain reaction (PCR). This stage enriches the cleaved fragments, which now harbor information pertaining to both target and off-target cleavage sites.
Next-Generation Sequencing (NGS): The prepared library undergoes NGS, yielding millions of reads corresponding to the DNA fragments cleaved by the nuclease.
Bioinformatics Analysis: The sequencing data undergo scrutiny via specialized bioinformatics pipelines. Tools such as BWA and SAMtools facilitate alignment of the reads to a reference genome, enabling identification of DSBs at both on-target and off-target loci.
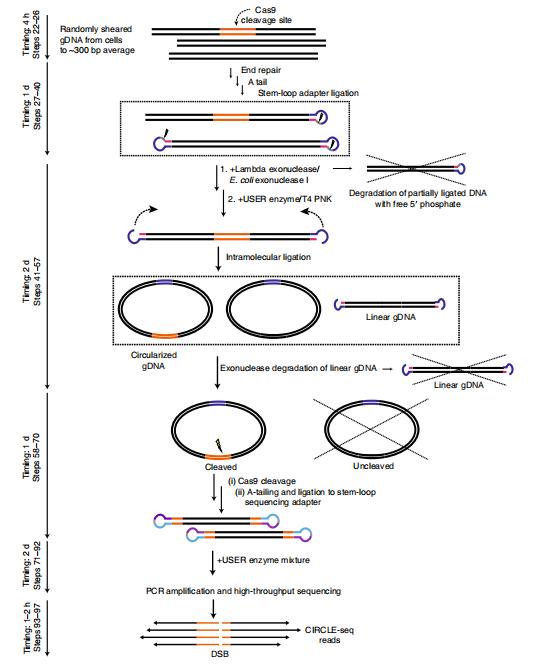
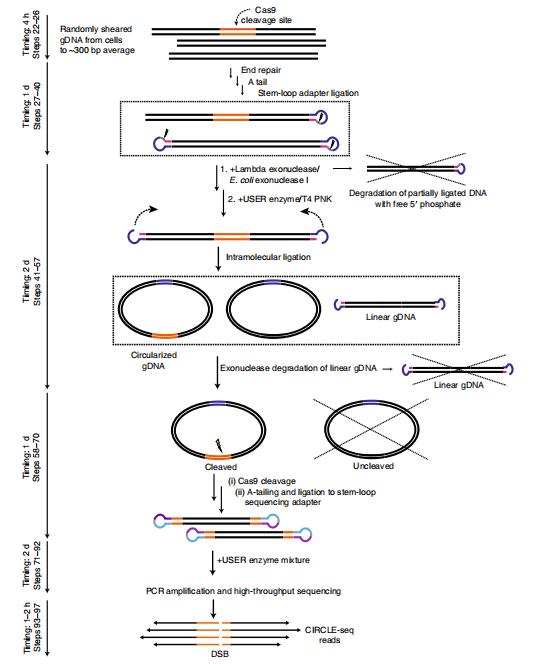 Overview of CIRCLE-seq workflow.
Overview of CIRCLE-seq workflow.
Circle-Seq Analysis
Efficiency and Error Correction
Tools for extrachromosomal DNA (ecDNA) research have advanced notably with the advent of Circle-seq methodology. Circle-seq, distinguished by its efficacy and error-correction capabilities when juxtaposed with conventional barcoding approaches, stands as a cornerstone in contemporary molecular investigations.
Efficiency, denoting the ratio of consensus bases generated to the total bases utilized, constitutes a pivotal metric in assessing sequencing methodologies. Under ideal conditions, wherein read families uniformly comprise three members, an efficiency benchmark of 33% is attainable. However, pragmatic considerations, encompassing the variance in circular DNA lengths and the employment of paired-end reads, lead to a marginally diminished efficiency.
Notably, CD Genomics has achieved an efficiency milestone of 20.2% in Circle-seq applications. This achievement signifies the derivation of one consensus base for every five bases engaged in sequencing endeavors. Contrastingly, conventional barcoding techniques typically exhibit efficiencies within the range of 1-8%. For instance, standard barcoding and duplex barcoding methodologies yield consensus sequences with efficiencies of 3.0% and 0.8%, respectively.
The superior efficiency characteristic of Circle-seq not only engenders the acquisition of enhanced quality data but also underpins a reduction in overall investigative costs. Thus, Circle-seq emerges as an indispensable tool in the arsenal ecDNA researchers, heralding a paradigm shift in molecular exploration.
Robustness Across Experimental Conditions
Circle sequencing stands out as a formidable tool owing to its resilience under diverse experimental conditions. Unlike barcoding methodologies, which necessitate meticulous manipulation of the ratio between barcoded input molecules and total reads, circle sequencing exhibits steadfast efficiency irrespective of the scale of the input library or the magnitude of generated reads. This exceptional robustness stems from the inherent physical linkage of reiterated sequences within individual read families, thus obviating the inherent variability associated with sampling from a complex mixture. Such intrinsic stability elevates circle sequencing as a reliable choice for molecular investigations across a spectrum of research settings.
Applications in Genomic Research
Circle sequencing emerges as a pivotal tool in scenarios demanding unparalleled accuracy and efficacy. In the realm of cancer profiling, for instance, the method’s capacity to furnish high-fidelity, error-corrected datasets assumes critical significance in discerning rare mutations and elucidating tumor heterogeneity. Likewise, within the domain of immunogenetics, circle sequencing offers a conduit for probing the intricacies of immune receptor repertoires, thereby illuminating immune responses and underlying disease mechanisms.
Moreover, circle sequencing finds resonance in microbial diversity investigations and environmental sampling endeavors. The method’s exceptional efficiency and resilience facilitate the identification and characterization of elusive microbial species and intricate microbial communities, which often elude detection by conventional sequencing modalities. This multifaceted utility positions circle sequencing as a cornerstone in molecular explorations spanning diverse disciplines.
Circle-seq Bioinformatics Steps
1. Read Mapping and Alignment
The sequence reads produced via Circle-seq experiments are scrutinized, positioned, and coordinated with the reference genome via application of specialized bioinformatics tools like the Burrows-Wheeler Aligner (BWA) and Sequence Alignment/Map (SAMtools). This crucial step facilitates the precise identification and subsequent classification of circular DNA elements within the genome’s intricate architecture, thus ensuring a deeper understanding of our genetic fabric.
2. Identification of Circular DNA Elements
Circular DNA motifs are discerned according to their distinct alignment blueprints and distribution of sequence reads. Employing refined bioinformatics algorithms serves to set apart circular DNA elements from their linear genomic counterparts, thereby enabling accurate positioning and intricate profiling of circular genomic structures.
3. Functional Annotation and Interpretation
Given the identification of the circular DNA elements, the task then shifts to their functional annotation and interpretation to shed light on their biological relevance. This consists of associating the circular DNA elements with distinct genomic features, for instance, gene promoters, enhancers, and regulatory elements, which yields insights into their probable roles in gene regulation and genome stability.
Advantages of Circle-seq
Circle-seq has emerged as an instrumental molecular technique, boasting significant advantages over classical sequencing methods, largely owing to its capacity to enrich circular DNA molecules selectively. This capability, in turn, enables an in-depth exploration of genomic components and uncovers their functional implications.
1. Enhanced Sensitivity and Specificity
A salient advantage attributed to Circle-seq lies in its unparalleled sensitivity and specificity in pinpointing circular DNA constituents within complex genomic landscapes. Leveraging a selective enrichment approach for circular DNA fragments, Circle-Seq mitigates confounding signals and false-positive results, thus augmenting the accuracy of genomic profiling. The technology’s heightened specificity ensures the reliable detection of circular DNA elements, transcending the limitations imposed by scarce abundance. Consequently, Circle-Seq is especially appropriate for the investigation of infrequent genomic events and structural aberrations.
2. Comprehensive Genomic Coverage
Circle-seq stands as an instrumental technology in the domain of molecular biology, providing an inclusive genomic coverage, and equipping scientists with the capacity to identify various circular DNA elements. These include ecDNA, circRNA, and a plethora of other circular genomic fragments. In contrast to conventional sequencing methodologies, which primarily lay emphasis on linear DNA trajectories, Circle-seq extends a comprehensive perspective on the genomic circular landscape. This not only maps the genomic architecture in its totality but also facilitates unearthing of previously unidentified, integral insights on genomic organization.
3. Versatility and Flexibility
Paralleling a considerable asset of the Circle-seq methodology is its versatile deployment potential in genomic studies. Exhibiting adaptability to an array of experimental designs and sample typologies, Circle-seq is aptly suited to address a broad range of research queries and targets. Its application extends from the examination of oncological molecular processes, the scrutiny of chromatin topography to the regulatory analysis of RNA. Further elevating Circle-seq’s versatility is its potential integration with other molecular methodologies. Integration with techniques such as chromatin immunoprecipitation (ChIP) and RNA sequencing (RNA-seq) harnesses the opportunity to glean additional perspectives into genomic function and its regulation mechanisms.
4. Cost-effectiveness
Incorporating both technical and economic efficiency, Circle-seq distinguishes itself from more conventional sequencing methods. Its selective enrichment of circular DNA molecules curtails the need for profound sequencing depth to achieve exhaustive genomic coverage. This, in turn, effectively mitigates the overall expenses associated with sequencing. Consequently, Circle-seq emerges as a viable and cost-effective solution for extensive genomic research activities and analyses involving numerous samples. Such efficient financial management could indeed be invaluable in contexts where budgetary constraints pose a significant challenge to the use of high-priced sequencing methodologies.
Comparison to Other Methods
While Circle-seq offers numerous advantages, it is essential to compare this technique to other sequencing methods to understand its relative strengths and limitations.
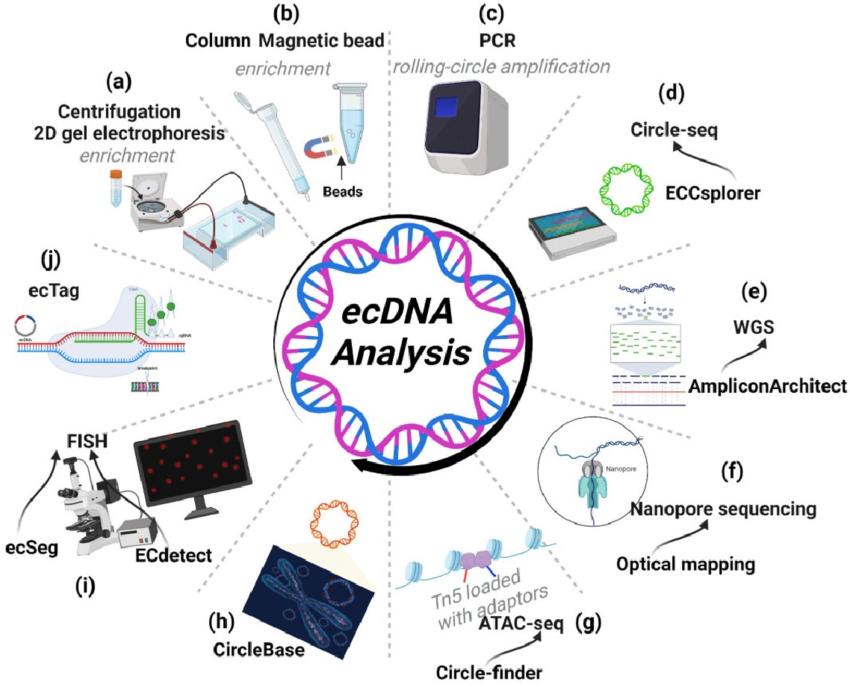
Tools for ecDNA research. The study of ecDNA is enriched by several sophisticated tools, among which Circle-seq figures prominently. The workflow of Circle-seq initiates with an enrichment protocol that employs either density gradient centrifugation or 2D gel electrophoresis, followed by the utilization of column- or bead-based enrichment methods. Prior to sequencing, rolling-circle amplification is employed to enhance the available ecDNA (A-C). Upon completing Circle-seq, ECCsplorer serves as a valuable tool for swift and efficient identification of potential ecDNA (D). WGS data can be utilized by AmpliconArchitect to extract valuable ecDNA information, offering an alternative avenue for data derivation (E). For discerning complex ecDNA architectures, nanopore long-read sequencing and optical mapping offer unmatched detail (F). ATAC-seq, in conjunction with Circle-finder software, serves as yet another potential route for ecDNA identification in cancer tissues (G). CircleBase, a comprehensive platform for integrating and analyzing ecDNA resources, enables screening for functional ecDNAs and offers insights into their molecular mechanisms (H). Fluorescence In Situ Hybridization (FISH) allows gene amplification on ecDNA to be identified, with ECdetect serving as a novel, integrated analysis pipeline for ecDNA quantification from metaphase cells stained with DNA stains like DAPI. The efficacy and sensitivity of detecting DAPI-stained ecDNAs in metaphase cells can be further augmented by ecSeg, which employs a deep neural network (I). Lastly, ecTag provides a unique tool for labeling ecDNA with multiple fluorescent molecules in living cells (J).
1. Comparison to Linear DNA Sequencing
Serving as an innovative technique in the realm of molecular biology, Circle-seq differentiates itself from conventional linear DNA sequencing methods that centralize primarily on the linear DNA sequences—precisely, Whole Genome Sequencing (WGS) and Targeted Amplicon Sequencing. Unlike these traditional methodologies, Circle-seq directs its focus explicitly on circular DNA molecules, thereby offering a more inclusive perspective of the genome’s circular architecture. This distinguishing proficiency for capturing circular DNA elements sets Circle-seq apart from the conventional linear DNA sequencing methodologies. Consequently, it facilitates the detection of distinct genomic characteristics, such as ecDNA and circRNA, which may otherwise remain obscured when employing traditional sequencing practices.
2. Comparison to Chromatin Conformation Capture (3C) Techniques
Whilst Chromatin Conformation Capture (3C) techniques such as Hi-C and ChIA-PET have been favourably employed to analyze chromatin architecture and long-range chromatin interactions, these techniques primarily focus on linear DNA sequences, often disregarding circular DNA elements. On the contrary, Circle-seq specifically enriches for circular DNA molecules, facilitating a comprehensive profiling of circular genomic features. This focused approach enables Circle-seq to effectively study ecDNA and other circularized genomic fragments, which are critical to genome organization and function.
3. Comparison to RNA-seq for circRNA Detection
RNA-seq, a method ubiquitously employed for transcriptomic interrogation, encompasses the task of identifying circRNA molecules. While RNA-seq affords indispensable knowledge on gene expression and alternative splicing events, its proficiency in accurately detecting circRNA is considerably hindered, constrained by factors inclusive of RNA fragmentation methodologies and bioinformatics computation algorithms.
In notable deviation, Circle-seq operates as an alternative, utilizing a targeted strategy for circRNA identification. This method garners an enrichment of circular RNA molecules specifically, thereby endowing heightened sensitivity and specificity compared to the foundational RNA-seq approach. Circle-seq’s distinguished ability to selectively ensnare circRNA molecules furnishes it as a crucial asset in the pursuit of in-depth understanding regarding circRNA biogenesis, regulation mechanisms, and the role of circRNA in the orchestration of diverse biological processes.
In conclusion, Circle-seq exhibits multiple strengths over conventional sequencing modalities, constituted by superior sensitivity, extensive genomic coverage, adaptability, and cost efficacy. By judiciously enriching for circular DNA fragments, Circle-seq facilitates an accurate delineation and elucidation of circular genomic structures, dispensing critical understanding of genome organization, regulation, and functionality. Moreover, in juxtaposition with alternative sequencing methods, Circle-seq distinguishes itself via its unique capacity to encapsulate singular circular DNA constituents, facilitating the revelation of previously unearthed dimensions in genome biology.
Applications of Circle-seq
In essence, Circle-seq manifests as a pioneering approach that has been instrumental across various facets within the realm of genomic investigation, especially in augmenting both accuracy and safety parameters inherent in genome modification processes. Circle-seq facilitates precise identification of any peripheral, unintended implications (off-target effects), thus buttressing a spectrum of scholarly explorations and potential therapeutic interventions. Subsequent sections delve into an assortment of critical applications of Circle-seq, underscored by empirical instances culled from scientific literature.
Detecting Off-Target Effects in CRISPR-Cas9 Editing
Comprehensive Mapping of CRISPR-Cas9 Off-Targets
In a seminal study by Tsai and colleagues (2017), they employed Circle-seq as a tool to systematically catalogue the off-target locations affected by the action of CRISPR-Cas9 nucleases. This examination underscores that Circle-seq may reveal a more extensive array of off-target mutations than other comparative methods, such as GUIDE-seq or Digenome-seq. The investigative team used Circle-seq on a diverse array of CRISPR-Cas9 constructs, revealing noteworthy differences in off-target activity. These findings underscore the utility of Circle-seq in the refinement of genome editing tools, illuminating its potential application within clinical domains.
Reducing Off-Target Effects in Therapeutic Applications
In the domain of gene therapy, safeguarding the accuracy of genome editing is a top priority; this ensures the elimination of inadvertent genetic modifications capable of triggering deleterious consequences. In pursuit of this aim, an investigation presented in “Nature Biotechnology” by Kosicki and his team in 2018 leveraged the Circle-seq technique with the objective of scrutinizing the off-target ramifications of CRISPR-Cas9 within the realm of human hematopoietic stem cells. Their results indicated that Circle-seq held the capacity to discern infrequent off-target occurrences, which noncomparably evaded detection by alternative methodologies, thereby offering a more all-encompassing safety overview for prospective therapeutic strategies.
Assessing Genome Integrity in Edited Cells
In the interest of preserving genome stability in a plethora of biotechnological and therapeutic contexts, a study by Wienert et al. (2019), featured in “Nature Communications,” engaged the use of Circle-seq to examine genome constancy within cells edited using CRISPR-Cas9. Their findings suggested that Circle-seq was proficient in the detection of intricate genomic rearrangements in addition to large-scale deletions manifesting at sites of off-target activity. Such an application underscores the critical role of Circle-seq in safeguarding the enduring safety of edited cells, especially those intended for utilization in regenerative medicine and cell-based therapies.
Comparison of Genome-Editing Nucleases
Ameliorating the precision of genome-editing tools is an ongoing challenge in the realm of biological sciences. In this regard, Circle-seq has emerged as a superior method to assess the off-target activities of a plethora of genome-editing nucleases. Well-documented by the study of Kim et al. (2019) appearing in the scientific journal, “Genome Research”; this investigation juxtaposed the off-target impacts of varied CRISPR-Cas variants inclusive of Cas9, Cas12a (Cpf1), and Cas13. The study accentuated the remarkable capability of Circle-seq to delve deeper into the nuances of the specificity of each nuclease, thereby facilitating the evolution of subsequent generation genome-editing instruments with enhanced precision.